3.3. Peak-calling
In the PC_SHARP and PC_BROAD modes, drompa+ can call peaks by supplying the --callpeak option:
$ dir=parse2wigdir+
$ drompa+ PC_SHARP \
-i $dir/H3K4me3.100.bw,$dir/Input.100.bw,H3K4me3,,,100 \
-i $dir/H3K27me3.100.bw,$dir/Input.100.bw,H3K27me3,,,10 \
-i $dir/H3K36me3.100.bw,$dir/Input.100.bw,H3K36me3,,,10 \
-o drompa4 -g refFlat.txt --gt genometable.txt \
--lpp 2 --showitag 2 --callpeak
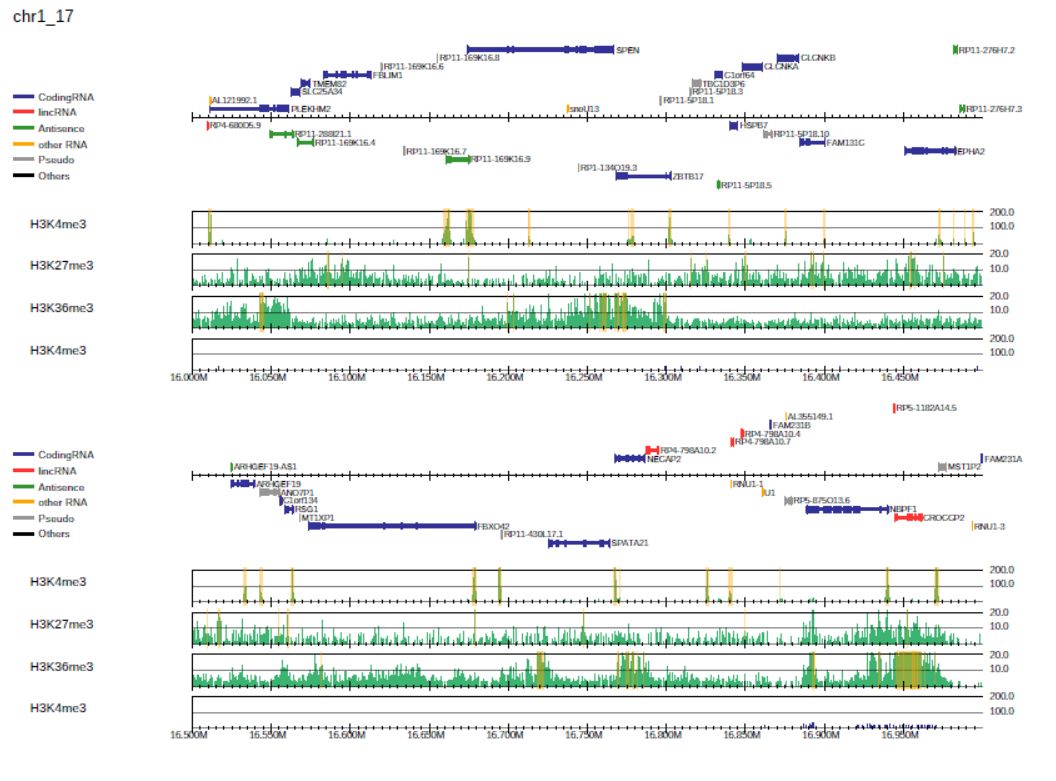
Fig. 3.18 Highlighting peaks.
Peak regions are highlighted in orange. The peak list for each sample pair is also outputted as “<output-name>.<label>.peak.tsv”:
$ ls drompa4.*.tsv
drompa4.H3K27me3.peak.tsv drompa4.H3K36me3.peak.tsv drompa4.H3K4me3.peak.tsv
The peak list is in tab-delimited text format and can be opened in a text editor or Microsoft Excel. It contains the following columns:
chromosome name
start position
end position
peak summit (center of summit bin)
peak width
ChIP read pileup
Input read pileup
ChIP/Input enrichment (pileup)
P-value (ChIP internal)
P-value (ChIP/Input enrichment)
peak name.
Note
If a BED file is specified using the
-ioption, drompa+ does not internally call peaks but highlights the specified regions instead.By default, chromosomes Y and M (Mt) are ignored during analysis. Supply the
--includeYMoption to include these chromosomes.When supplying the
--chroption, peaks for only the specified chromosome are called.Supply the
--offpdfoption to omit PDF file generation and obtain peak lists only.
3.3.1. Detail of significance test
The PC_SHARP and PC_BROAD modes adopt a two-step procedure for the significance testing of peak-calling.
In the first step, they identify significantly enriched sites compared to a background null model, assuming a Poisson distribution in local background region (100 kbp).
In the second step, from these candidate sites, they identify significantly enriched ones compared to the input control based on the binomial distribution.
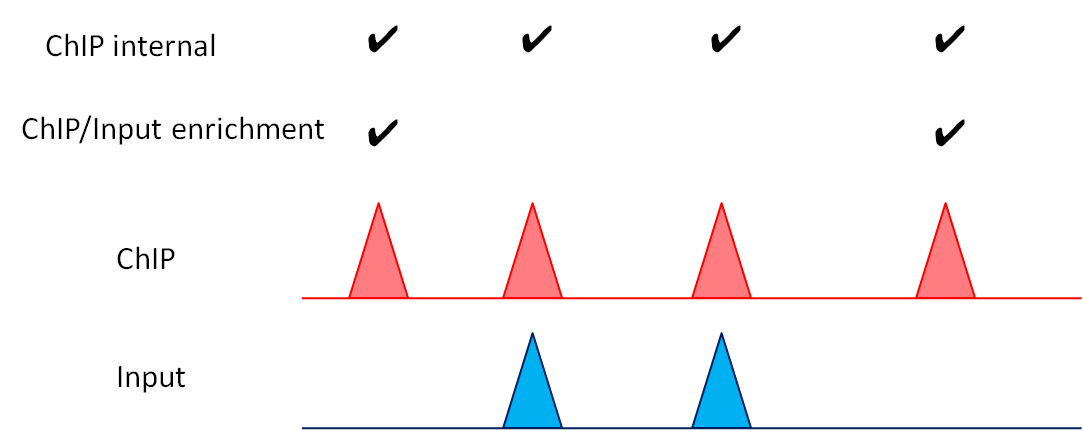
Fig. 3.19 Schematic representation of the peak-calling thresholds.
Accordingly, there are multiple thresholds for peak-calling, as discussed below:
Main thresholds:
--pthre_internal: the p-value of the first step (ChIP-internal enrichment)--pthre_enrich: the p-value of the second step (ChIP/Input enrichment)
Optional thresholds:
--ethre: the ChIP/Input enrichment--ipm: the normalized intensity (height) of the peak summit--nbin4lmd: the number of bins for the local Poisson (--pthre_internal). If the chromosome size is smaller than the specified length, the entire chromosome is used.
See --help for the default threshold values for each drompa+ mode.
The --pthre_enrich option is recommended as the main threshold for peak-calling.
3.3.2. Peak-calling without the input sample
If the input sample is not specified, drompa+ calls peaks using the ChIP sample (--pthre_internal) and skips the second step (--pthre_enrich) of the peak-calling procedure.
However, we strongly recommend that the ChIP sample is compared with the corresponding input data to decrease the number of false positive sites derived from repeated regions.
3.3.3. Peak-calling in PC_ENRICH mode
By default, the PC_ENRICH mode does not implement a significance test but simply calls regions containing ChIP/Input enrichments above the enrichment threshold (--ethre, 2.0 by default) and the peak intensity threshold (--ipm, 5.0 by default).