DROMPAplus: a pipeline tool for ChIP-seq analysis
DROMPA (DRaw and Observe Multiple enrichment Profiles and Annotation) is a ChIP-seq pipeline tool that meets various needs, including quality check, analysis, and visualization of multiple ChIP samples.
DROMPAplus is an update of DROMPA3. It is written in C++ and runs from a single launch command on conventional Linux systems.
The main features of DROMPAplus are summarized below. DROMPAplus:
Accepts multiple map file formats (SAM, BAM, CRAM, Bowtie, TagAlign(.gz)) and read distribution formats (WIG(.gz), bigWig, bedGraph).
Supports spike-in normalization and total read normalization.
Outputs various quality metrics for ChIP-seq analysis.
Visualizes read distributions in conventional PDF format; therefore, no additional programs are required which is preferable for many users, especially when sharing results (e.g., on cloud storage) with collaborators who do not have a strong bioinformatics background.
Automatically estimates the fragment length from single-end reads using SSP.
Can visualize two samples in a single line, which delineates the co-occurrence (e.g., H3K4me3 and H3K27ac) and exclusivity (e.g., H3K27me3 and H3K36me3) of read enrichment. Transparency (alpha) of read color can be specified.
Supports chromatin loops from ChIA-PET (Mango format) and Hi-C (HICCUPS format) with colors corresponding to the p-values.
Bases the HEATMAP command on Python3, which enables flexible customization.
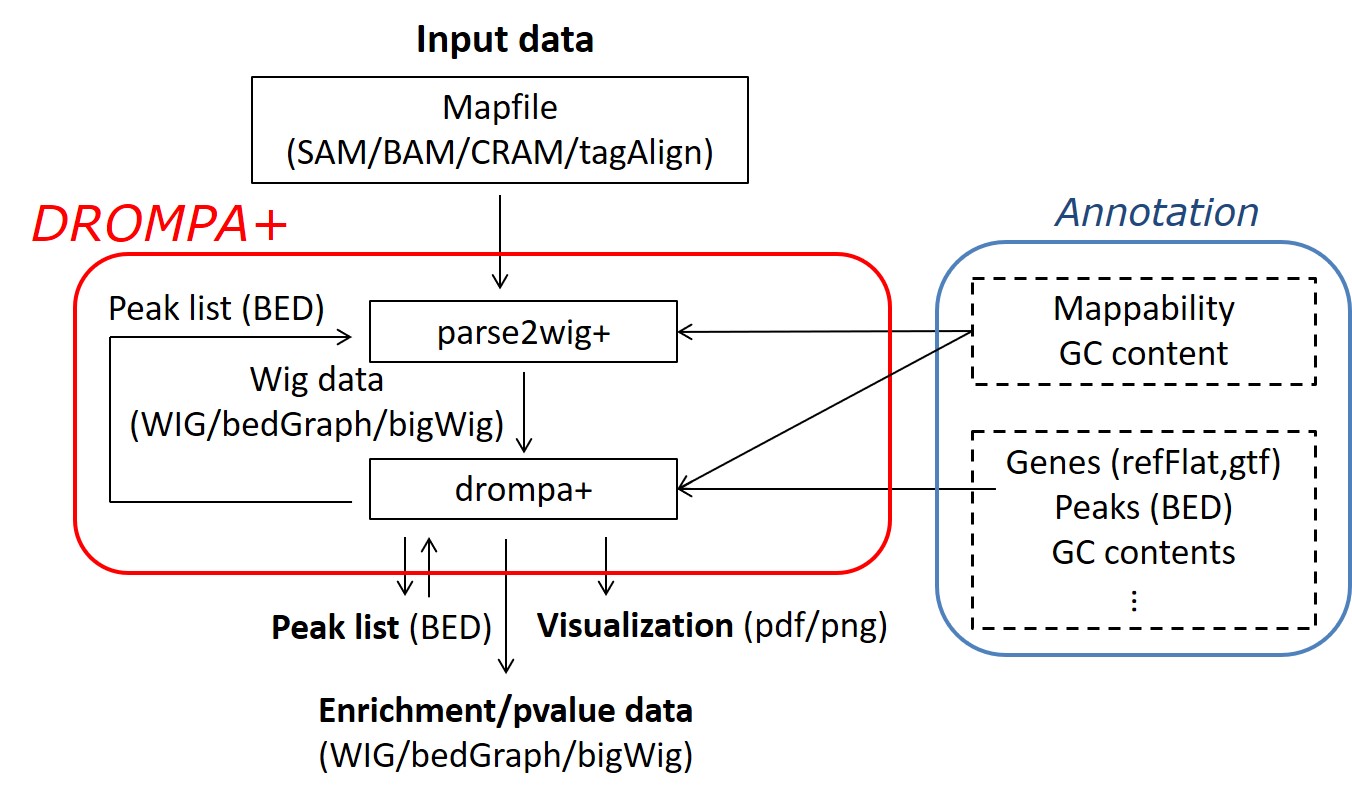
Fig. 1 Schematic representation of DROMPA+ features and functionality.
Contents:
Citation:
Nakato R., Sakata T., Methods for ChIP-seq analysis: A practical workflow and advanced applications, Methods, 2020.
Contact:
- Mail:
rnakato AT iqb.u-tokyo.ac.jp
- Twitter:
@RyuichiroNakato